






A new, freely available AI catalog has classified the potential effects of millions of missense genetic mutations, which could help establish the cause of diseases such as cystic fibrosis, sickle-cell anemia, and cancer.
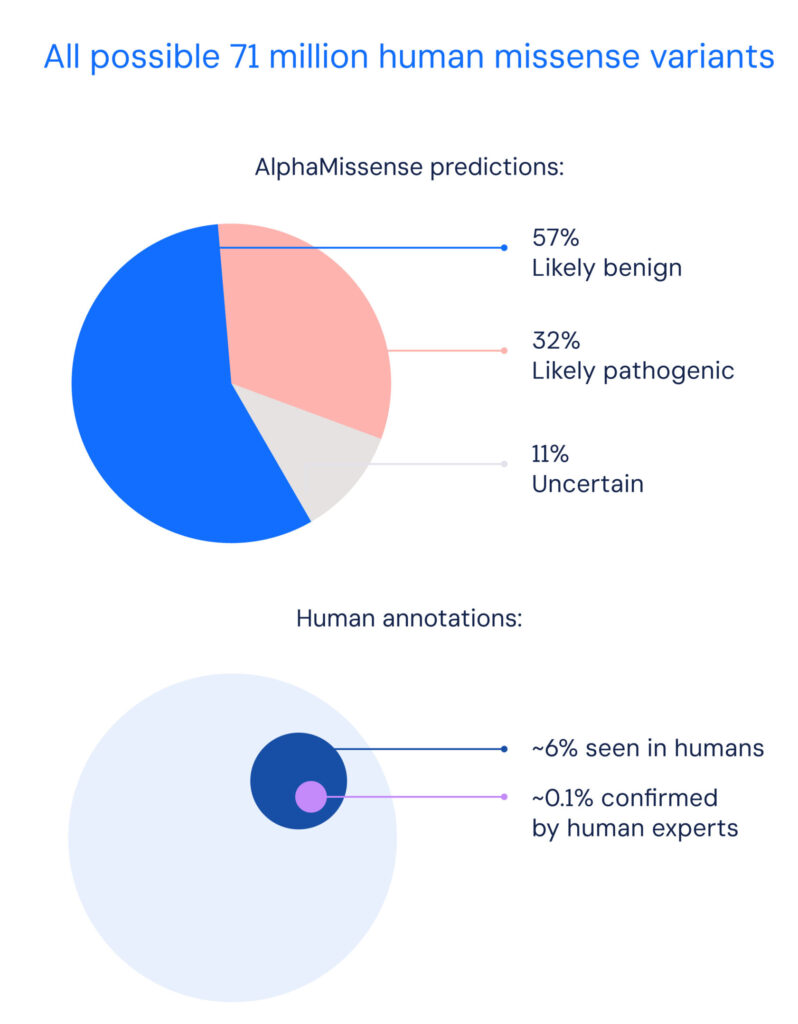
The AlphaMissense resource from Google DeepMind categorized 89% of all 71 million possible missense variants.
This compares with just 0.1% that have been already categorized by human experts.
The machine-learning algorithm predicted 57% as likely benign and 32% likely highly pathogenic, using a threshold that yielded 90% precision on a database of known disease variants.
“AlphaMissense achieves state-of-the-art predictions across a wide range of genetic and experimental benchmarks, all without explicitly training on such data,” Jun Cheng and colleagues at Google Deepmind assert in a blog accompanying their findings in the journal Science.
Missense genetic mutations arise from a single letter substitution in DNA, resulting in an altered amino acid that can potentially affect the entire function of a protein.
The average person contains 9000 missense mutations, most of which are benign, and it remains largely a mystery which give rise to disease.
In some cases, a disease may result from single or few missense variants while other complex diseases such as Type 2 diabetes may result from a combination of many different types of genetic changes.
AI tools provide an alternative to expensive and laborious experiments, enabling researchers to gain a preview of results for thousands of proteins, allowing them to prioritize resources and fast-track more complex studies into their predicted effects.
The benefits could potentially cover fields ranging from molecular biology to clinical and statistical genetics.
The AlphaMissense resource is adapted from AlphaFold, a previously published breakthrough model that predicted the structures of almost all known proteins from their amino acid sequences.
The newly released variant effect predictor (VEP) algorithm predicts the pathogenicity of missense variants altering individual amino acids of proteins.
To train it, the team fine-tuned AlphaFold on labels that distinguished variants seen in humans and closely related primate populations from variants never seen in humans.
The catalog does not predict changes to protein structure from mutations or other effects on protein stability. Instead, it deploys databases of related protein sequences and the structural context of variants to score the likelihood of a variant being pathogenic on a continuous scale from zero to one.
This enables a threshold for classifying variants as pathogenic or benign to be chosen to match user accuracy requirements.
Together with EMBL’s European Bioinformatics Institute, the creators are making the information more usable for researchers through the Ensembl Variant Effect Predictor.
In addition to a look-up table of missense mutations, they have shared expanded predictions of all possible 216 million single amino acid sequence substitutions across more than 19,000 human proteins.
They have also included the average prediction for each gene, which is similar to measuring a gene’s evolutionary constraint—how essential the gene is for the organism’s survival.
“Our tool outperformed other computational methods when used to classify variants from ClinVar, a public archive of data on the relationship between human variants and disease,” the investigators note.
“Our model was also the most accurate method for predicting results from the lab, which shows it is consistent with different ways of measuring pathogenicity.”






